"El gen" de las enfermedades complejas
El gen de la esquizofrenia, el gen del lenguaje, el gen de la diabetes… ¡Incluso el gen de la infidelidad! ¿Cuántas veces hemos visto cómo los titulares reducen erróneamente enfermedades o rasgos humanos complejos a un único gen? En la mayoría de los rasgos y enfermedades comunes, intervienen más de un gen y además, los factores ambientales juegan un papel importante. Las enfermedades complejas o multifactoriales son aquellas enfermedades que están causadas por el efecto combinado de diferentes genes y en las que además influyen los factores ambientales de forma considerable, estas enfermedades son difíciles de estudiar, algunas de estas son la diabetes, la esquizofrenia, el alzhéimer, la psoriasis, la artritis reumatoide o el asma.

La esquizofrenia es una enfermedad compleja considerada un trastorno psiquiátrico grave que afecta al 1% de la población, los síntomas más comunes en la esquizofrenia son delirios, alucinaciones, alteraciones en el pensamiento, afectividad, conducta y cognición. Esta enfermedad muestra agregación familiar, es decir, los miembros de una familia tienen mayor tendencia a desarrollar el mismo rasgo o enfermedad porque al margen de compartir una gran proporción de sus genes comparten el ambiente donde viven; sin embargo cuando se calcula el riesgo de una persona de sufrir esquizofrenia los principales determinantes de que esta persona tenga o no la enfermedad serán los genes, por lo que cuanto mayor DNA se comparta existe mayor probabilidad de que si una persona tiene esquizofrenia, la otra persona también tenga este trastorno sim embargo el ambiente también interviene relevantemente, para calcular la influencia de los genes en esta enfermedad se utiliza la heredabilidad, que en el caso de esta enfermedad es muy alta, alrededor del 81%. El gen DISC1 fue el primer gen encontrado relacionado con la esquizofrenia, aunque la presencia de alteraciones en el gen DISC1 no es garantía de desarrollar la esquizofrenia, sí aumenta considerablemente el riesgo; DISC1 regula el esqueleto interno de la célula y podría contribuir a la esquizofrenia alterando las funciones neuronales que dependen de este esqueleto, como por ejemplo la migración, el transporte intracelular o la arquitectura de las prolongaciones nerviosas. Gracias a los estudios de asociación del genoma completo (o GWAS) e han encontrado asociaciones significativas entre variantes genéticas comunes y raras, aun así, encontrar una asociación significativa entre una variante genética y una enfermedad, no prueba que el gen donde se encuentra esté relacionado con la enfermedad; el primer gen identificado en un GWAS fue es el gen responsable de codificar el componente 4 del complemento, molécula que forma parte del sistema inmunitario innato y participa en la eliminación de patógenos y reciclaje de restos celulares. el gen se expresa de forma diferente en pacientes y controles y participa en el reciclaje de sinapsis nerviosas. El caso de la esquizofrenia es sólo un ejemplo, cada vez se conoce mejor el genoma y existen más herramientas para su análisis y así se pueden describir mejor estas enfermedades complejas, todos estos conocimientos sin olvidar los factores ambientales serán claves para diseñar estrategias terapéuticas frente a estas enfermedades.
Noticia
https://revistageneticamedica.com/blog/gen-las-enfermedades-complejas/ Bibliografía
Birnbaum R, Weinberger DR. Genetic insights into the neurodevelopmental origins of schizophrenia. Nat Rev Neurosci. 2017 Dec;18(12):727-740. doi: 10.1038/nrn.2017.125.
Kotlar AV, et al. New discoveries in schizophrenia genetics reveal neurobiological pathways: A review of recent findings. Eur J Med Genet. 2015 Dec;58(12):704-14. doi: 10.1016/j.ejmg.2015.10.008. Sekar A, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016. doi: 10.1038/nature16549
Samson JN y Wong AHC. The Genetics of Schizophrenia. Drug Discovery for Schizophrenia. Doi: http://dx.doi.org/10.1039/9781782622499-00001
By Antonio Sánchez (DGMol)
Los científicos se encuentran genes detrás de una enfermedad cardíaca mortal
Identificación de la rara variación de secuencia subyacente a la hipertensión arterial pulmonar hereditaria.
La hipertensión arterial pulmonar (HAP) es un trastorno hereditario, en la mayoría de los casos, poco frecuente, caracterizado por la oclusión de arteriolas en el pulmón lo que suele provocar la muerte a los 3 o 5 años después del diagnóstico en el 50% de los afectados. Hasta ahora se sabía muy poco de las causas que provocaban esta enfermedad, pero gracias al trabajo de un equipo de investigación británico, se han descubierto cuatro genes que hacen que se desarrolle la enfermedad: GDF2, ATP13A3, AQP1 y SOX17; mutaciones en estos genes producen un fallo en la ruta de señalización de TGF-β.
 El objetivo del estudio era conocer e identificar la variación adicional de secuencias raras que afectan a HPA y evaluar la contribución relativa de las diversas mutaciones. Este enfoque implicó una comparación rigurosa de casos y controles utilizando una búsqueda escalonada de variantes. Se compararon más de 1000 pacientes con HAP con un grupo control de más 6000 personas que no padecían la enfermedad. En general, para las mutaciones muy raras, la comparación entre los casos de los pacientes con HAP y los controles no debe reducir la capacidad de detectar la sobreexpresión estas en un gen particular, si las mutaciones son específicas de HAP. Sin embargo, si las mutaciones en un gen fueran responsables de más de un fenotipo, es posible que se reduzca la capacidad de detectar otros genes implicados en la enfermedad.
En un futuro cercano, este hallazgo facilitaría el diagnostico de las personas afectadas y en uno un poco más lejano, podría suponer un cambio en el tratamiento, que actualmente consiste en realizarle al paciente un trasplante de corazón o de pulmón.
El objetivo del estudio era conocer e identificar la variación adicional de secuencias raras que afectan a HPA y evaluar la contribución relativa de las diversas mutaciones. Este enfoque implicó una comparación rigurosa de casos y controles utilizando una búsqueda escalonada de variantes. Se compararon más de 1000 pacientes con HAP con un grupo control de más 6000 personas que no padecían la enfermedad. En general, para las mutaciones muy raras, la comparación entre los casos de los pacientes con HAP y los controles no debe reducir la capacidad de detectar la sobreexpresión estas en un gen particular, si las mutaciones son específicas de HAP. Sin embargo, si las mutaciones en un gen fueran responsables de más de un fenotipo, es posible que se reduzca la capacidad de detectar otros genes implicados en la enfermedad.
En un futuro cercano, este hallazgo facilitaría el diagnostico de las personas afectadas y en uno un poco más lejano, podría suponer un cambio en el tratamiento, que actualmente consiste en realizarle al paciente un trasplante de corazón o de pulmón.
Noticia
http://www.bbc.com/news/health-43727026
Referencia
https://www.nature.com/articles/s41467-018-03672-4#Abs1
Stefan Gräf et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nature Communications. Springer Nature. Apr 12, 2018. 1416 (2018) doi:10.1038/s41467-018-03672-4
By María Bohoyo (DGMol)
POR QUÉ NUESTROS HIJOS PODRÍAN HEREDAR NUESTROS VICIOS
Sabemos que en el interior de nuestras células está nuestro genoma, el DNA, en el esta “registrada” toda nuestra información genética heredada al azar de nuestros padres. Cada vez más se conoce otro concepto muy relacionado con este que es el de epigenoma. Nuestro epigenoma son todas aquellas modificaciones (Ej. metilaciones) que “marcan” nuestro genoma, estas son producidas por cambios en nuestra alimentación, condiciones ambientales como puede ser la contaminación o como habla esta noticia el consumo de drogas.
 Desde hace tiempo se sabe que hay factores genéticos heredables que pueden provocar que una persona sea más propensa que otra a sufrir una adicción, estudios demuestran que el riesgo de abusar de las drogas es heredable hasta en un 60% de los casos. Como he comentando en la introducción a esta noticia se conoce que ciertas marcas químicas producidas por nuestros hábitos se agregan a nuestro genoma y estas podrían además heredarse a generaciones posteriores.
En un estudio realizado en una Universidad de Shanghái (China), utilizaron ratones para intentar demostrar si era cierto que, si un ratón tenía adicción a la droga sus descendientes iban a ser más propensos a tenerla. En este estudio separaron a ratones en función de su motivación para buscar la droga, una vez identificaron aquellos con más interés por tomar cocaína observaron la posible relación que tenían las crías con esta sustancia y observaron que tenían más posibilidades de engancharse.
Para entender mejor los factores que podían hacer heredable esta adicción, los
investigadores compararon el esperma de los ratones con más interés por consumir y el de los que no tenían interés, y encontraron diferencias de metilación en algunas zonas del DNA. Estas metilaciones son los cambios epigenéticos heredables que cambiaban la expresión de los genes en un individuo u otro.
Aunque este estudio no ha sido realizado en humanos, nos deja evidencias y debería hacernos cambiar de opinión en algunos aspectos de nuestra vida, sobretodo a la hora de elegir nuestros hábitos porque no solo estaremos perjudicándonos a nosotros mismos, si no también a nuestros futuros hijos, y nietos.
Desde hace tiempo se sabe que hay factores genéticos heredables que pueden provocar que una persona sea más propensa que otra a sufrir una adicción, estudios demuestran que el riesgo de abusar de las drogas es heredable hasta en un 60% de los casos. Como he comentando en la introducción a esta noticia se conoce que ciertas marcas químicas producidas por nuestros hábitos se agregan a nuestro genoma y estas podrían además heredarse a generaciones posteriores.
En un estudio realizado en una Universidad de Shanghái (China), utilizaron ratones para intentar demostrar si era cierto que, si un ratón tenía adicción a la droga sus descendientes iban a ser más propensos a tenerla. En este estudio separaron a ratones en función de su motivación para buscar la droga, una vez identificaron aquellos con más interés por tomar cocaína observaron la posible relación que tenían las crías con esta sustancia y observaron que tenían más posibilidades de engancharse.
Para entender mejor los factores que podían hacer heredable esta adicción, los
investigadores compararon el esperma de los ratones con más interés por consumir y el de los que no tenían interés, y encontraron diferencias de metilación en algunas zonas del DNA. Estas metilaciones son los cambios epigenéticos heredables que cambiaban la expresión de los genes en un individuo u otro.
Aunque este estudio no ha sido realizado en humanos, nos deja evidencias y debería hacernos cambiar de opinión en algunos aspectos de nuestra vida, sobretodo a la hora de elegir nuestros hábitos porque no solo estaremos perjudicándonos a nosotros mismos, si no también a nuestros futuros hijos, y nietos.
Noticia
https://elpais.com/elpais/2017/05/31/ciencia/1496244370_258847.html
Referencia
https://www.nature.com/articles/ncomms15527
By Andrea Herrera (GM)
Mutaciones en el gen SLC7A8 producen sordera asociada a la edad
 La presbiacusia es la pérdida auditiva ligada al envejecimiento. Uno de los síntomas más característicos de ésta es la reducción de la percepción de sonidos agudos. Sin embargo, ¿qué tiene que ver el gen SLC7A8 con la pérdida auditiva? Para responder a esta pregunta deberemos hablar antes del gen SLC7A8. Tras la expresión de este gen se forma el heterodímero SLC7A8/SLC3A2, el cual es un intercambiador de aminoácidos que se suele situar en la membrana plasmática de ciertos tipos celulares. Así, Meritxell Espino Guarch, Manuel Palacín y Virginia Nunes observaron que SLC7A8 se expresa en el oído interno de ratones y que su inactivación o desactivación da lugar a presbiacusia acelerada causada por daños en las estructuras de la cóclea.
La aparición de esta enfermedad se debe a que la falta de SLC7A8 da lugar a la degeneración del órgano de Corti, del ganglio espinal, de la estría vascular y del ligamiento espiral. Es por esto que SLC7A8 se ha relacionado con la pérdida de audición ligada al envejecimiento.
Además, gracias al estudio con pacientes con presbiacusia y otros controles les permitió identificar variantes del gen y se detectaron disminuciones significativas en la actividad de dicho gen en las variantes identificadas en los pacientes (algo que involucra aun más a SLC7A8 con el desarrollo de presbiacusia).
Pero ¿en qué medida afecta esto a la vida de los pacientes? La identificación de la relación de este gen con la presbiacusia podría ser de utilidad para identificar individuos con tendencia a desarrollar la enfermedad, lo que podría llevar a un tratamiento preventivo. Además, este estudio destaca los transportadores de aminoácidos como nuevo objetivo para estudiar trastornos en la audición.
La presbiacusia es la pérdida auditiva ligada al envejecimiento. Uno de los síntomas más característicos de ésta es la reducción de la percepción de sonidos agudos. Sin embargo, ¿qué tiene que ver el gen SLC7A8 con la pérdida auditiva? Para responder a esta pregunta deberemos hablar antes del gen SLC7A8. Tras la expresión de este gen se forma el heterodímero SLC7A8/SLC3A2, el cual es un intercambiador de aminoácidos que se suele situar en la membrana plasmática de ciertos tipos celulares. Así, Meritxell Espino Guarch, Manuel Palacín y Virginia Nunes observaron que SLC7A8 se expresa en el oído interno de ratones y que su inactivación o desactivación da lugar a presbiacusia acelerada causada por daños en las estructuras de la cóclea.
La aparición de esta enfermedad se debe a que la falta de SLC7A8 da lugar a la degeneración del órgano de Corti, del ganglio espinal, de la estría vascular y del ligamiento espiral. Es por esto que SLC7A8 se ha relacionado con la pérdida de audición ligada al envejecimiento.
Además, gracias al estudio con pacientes con presbiacusia y otros controles les permitió identificar variantes del gen y se detectaron disminuciones significativas en la actividad de dicho gen en las variantes identificadas en los pacientes (algo que involucra aun más a SLC7A8 con el desarrollo de presbiacusia).
Pero ¿en qué medida afecta esto a la vida de los pacientes? La identificación de la relación de este gen con la presbiacusia podría ser de utilidad para identificar individuos con tendencia a desarrollar la enfermedad, lo que podría llevar a un tratamiento preventivo. Además, este estudio destaca los transportadores de aminoácidos como nuevo objetivo para estudiar trastornos en la audición.
Noticia
https://revistageneticamedica.com/2018/04/04/presbiacusia-slc7a8/
Referencia
Espino Guarch M, et al. Mutations in L-type amino acid transporter-2 support SLC7A8 as a novel gene involved in age-related hearing loss. Elife. 2018 Jan 22;7. doi: http://dx.doi.org/10.7554/eLife.31511
By Rodrigo Martín (DGMol)
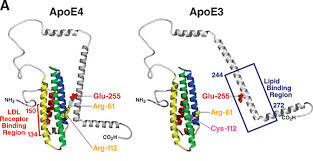
Desactivado el principal gen asociado al alzhéimer
Los investigadores de los Institutos de Gladstone en San Francisco, California (EE.UU.), han llevado a cabo una investigación pionera que muestra cómo uno de los factores de riesgo para el desarrollo de la enfermedad de Alzheimer crea ciertas señales en las células del cerebro. Estos investigadores no sólo se han limitado al estudio de dichas señales, sino que han logrado corregir el gen, eliminando los efectos nocivos.
Ya se sabía que el gen de la apolipoproteína (APOE) tenía un papel en el desarrollo del cáncer. Tener una copia de la variante de este gen triplica el riesgo de padecer Alzheimer y tener dos copias aumenta el riesgo en 12 veces más. APOE da lugar a lipoproteínas encargadas del transporte y regulación de los niveles de colesterol. La variante que lleva a esta enfermedad es la E4, la cual incrementa el riesgo de acumulación tóxica de beta amiloide y tau.
Lo que los investigadores buscaban era encontrar las diferencias entre las variantes E3 y E4. Para ello reprodujeron la enfermedad en células humanas y examinaron el efecto de APOE4 sobre las células del cerebro humano. Las neuronas se obtuvieron a partir de células de la piel de personas con alzhéimer que tenían dos copias de APOE4. También se utilizaron células de la piel de personas que no tenían Alzhéimer pero con dos copias de APOE3.
Observaron que en las células del cerebro humano APOE4 tiene una conformación patogénica que impide que funcione de manera adecuada, lo que lleva a la manifestación o desarrollo de diferentes enfermedades.
Otra observación destacable fue que encontraron que APOE4 aumenta la producción de beta-amiloide en humanos pero no en neuronas de ratones, lo que podría explicar las diferencias en ratones y humanos con respecto a la eficacia del fármaco.
Noticia
https://www.muyinteresante.es/salud/articulo/anulan-el-principal-gen-asociado-al-alzheimer-421523520614
Referencia
Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Chengzhong Wang, Ramsey Najm, Qin Xu, Dah-eun Jeong, David Walker, Maureen E. Balestra, Seo Yeon Yoon, Heidi Yuan, Gang Li, Zachary A. Miller, Bruce L. Miller, Mary J. Malloy & Yadong Huang. Nature Medicine (2018). DOI: doi:10.1038/s41591-018-0004-z
By Rodrigo Martín (DGMol)
La herencia del autismo de padres a hijos se relaciona con variantes
genéticas raras
Uno de cada 160 niños padece un trastorno del espectro autista (TEA), una patología que altera las habilidades comunicativas, el comportamiento social y el lenguaje.
La gran diversidad clínica y etiológica asociada a las patologías del TEA complica la definición de criterios diagnósticos y de prácticas clínicas y asistenciales. Sus causas son todavía bastante desconocidas. Sin embargo, los avances en genómica y biología molecular indican que tiene una base genética compleja. Un diagnóstico a tiempo es decisivo para iniciar intervenciones psicosociales desde el comienzo de la infancia y potenciar el progreso y el bienestar de las personas afectadas.
Un estudio internacional llevado a cabo con 2.600 familias revela el impacto de las variantes genéticas raras heredadas en el autismo, un trastorno poligénico y de difícil diagnóstico y tratamiento. La investigación, publicada en la revista Science, identifica diversas variantes relacionadas que están causadas por deleciones, duplicaciones en tándem, inversiones, cambios estructurales complejos y diversos tipos de elementos genéticos móviles que perfilan un nuevo paisaje genético para el espectro autista.
El estudio revela que las variantes estructurales identificadas contribuyen al 11 % de los casos de autismo, una cifra importante. De estas, la mitad son mutaciones de novo que afectan a genes concretos, y la otra mitad son mutaciones heredadas que alteran elementos reguladores o genes. Si a estas cifras se suman las mutaciones puntuales raras y las variaciones frecuentes
ya conocidas, estamos cada vez más cerca de perfilar el paisaje genético completo del espectro autista, aunque todavía queda bastante camino por recorrer. Lo novedoso de este trabajo es que explora el impacto en el autismo de las variantes genéticas raras heredadas, cuando la mayoría de trabajos anteriores investigan las variantes raras de novo, es decir, mutaciones que aparecen durante la formación de los gametos pero que no están presentes en los progenitores. El trabajo también se centra en las alteraciones
estructurales, es decir, en los cambios de segmentos largos de ADN, mientras que anteriormente la investigación analizaba sobre todo cambios de un único nucleótido.
Fundamento del estudio: El uso de la técnica de secuenciación del genoma entero (whole-genome sequencing, WGS) ha permitido identificar variantes estructurales que no eran detectables mediante otras técnicas por su reducido tamaño o por su localización en regiones no codificantes. WGS tiene la capacidad de secuenciar todo el genoma, pero que el impacto funcional de la mayoría de las alteraciones identificadas hasta ahora es todavía bastante desconocido Para resolver estas incertidumbres, son imprescindibles herramientas bioinformáticas sofisticadas análisis funcionales a gran escala.
En los pacientes, en concreto, se han estudiado regiones implicadas en la regulación de la expresión génica (inicios de transcripción, 3'-UTR y promotores del cerebro fetal) en genes que son intolerantes a las variantes estructurales (y que por tanto, cuando las tienen, es con consecuencias muy adversas).
Herencia: Por otro lado, el estudio revela que hay una mayor transmisión paterna de variantes estructurales en regiones reguladoras de la expresión génica que no toleran variaciones. Esto refuta la idea anterior de que el riesgo genético transmitido provendría predominantemente de las madres ya que se basa en su menor vulnerabilidad al trastorno.
Conclusiones
Los resultados de esta investigación ayudan a entender mejor los mecanismos genéticos que contribuyen a la aparición del autismo hereditario, y facilitarán el desarrollo futuro de herramientas de tratamiento farmacológico. El esfuerzo para definir con mayor precisión los componentes del paisaje genético del autismo ha dado lugar a un listado de genes que están alterados en los pacientes. Estos nuevos descubrimientos, que dan pistas sobre las grandes funciones del cerebro que están alteradas, también revelan cuáles son los elementos del genoma que podrían ser dianas para desarrollar futuras terapias.
La investigación revela que muchas de las alteraciones detectadas en los pacientes tienen un impacto sobre elementos que regulan la expresión de genes. Aunque aún se está lejos de las aplicaciones terapéuticas, esto podría abrir futuras opciones para obtener mejoras clínicas en los pacientes si somos capaces de modular —es decir, aumentar o disminuir— la expresión de determinados genes.
Noticia
http://www.agenciasinc.es/Noticias/
Referencia
William M. Brandler, Danny Antaki, Madhusudan Gujral, Morgan L. Kleiber, Joe Whitney et al."Paternally inherited cis-regulator ystructural variants are associatedwith autism". Science, 19 de abril de 201810.1126/science.aan2261
By Cristian Gragera (DG)
Un nuevo gen permite relacionar los defectos citoesquelétios con la esclerosis lateral amiotrófica (ELA)
ELA es una enfermedad neurodegenerativa que afecta a las neuronas implicadas en el movimiento. De esta forma, los afectados son incapaces de moverse y el destino final es la muerte. Esta enfermedad puede ser heredada (10%) o adquirida de novo (la mayoría).
 El grupo de investigación de Landers y Trainor ha conseguido relacionar el gen KIF5A con esta enfermedad. Los defectos en el citoesqueleto axónico en relación con la enfermedad permiten la búsqueda de nuevos fármacos enfocados en las proteínas del citoesqueleto.
Se llevó a cabo un estudio comparativo del genoma señalando este gen como uno de los implicados en la enfermedad. KIF5A codifica una proteína unida al citoesqueleto axónico y se encarga del movimiento y anclaje de estructuras. El déficit en esta proteína impide la correcta comunicación de las neuronas con las células post sinápticas.
Otras mutaciones que causan defectos en el citoesqueleto están relacionadas también con la ELA, confirmando éstas como una de las causas de la enfermedad. Además, alteraciones en dos rutas metabólicas también pueden ser causas de esta patología.
Este gen, ya había sido descrito como causante de otras dos enfermedades, paraplejia espástica hereditaria (HSP) y enfermedad de Charcot-Marie tipo 2 (CMT2). Sin embargo, y a diferencia con la ELA, las mutaciones se localizan en el extremo N-terminal (base de la proteína KIF5A), mientras que en la ELA, se encuentra en el C-terminal (extremo de unión a moléculas transportadoras de la proteína).
Por tanto, el siguiente objetivo es estudiar por qué una mutación en un extremo causa ELA y en el otro extremo causan HSP para así, desarrollar fármacos contra estas enfermedades.
El grupo de investigación de Landers y Trainor ha conseguido relacionar el gen KIF5A con esta enfermedad. Los defectos en el citoesqueleto axónico en relación con la enfermedad permiten la búsqueda de nuevos fármacos enfocados en las proteínas del citoesqueleto.
Se llevó a cabo un estudio comparativo del genoma señalando este gen como uno de los implicados en la enfermedad. KIF5A codifica una proteína unida al citoesqueleto axónico y se encarga del movimiento y anclaje de estructuras. El déficit en esta proteína impide la correcta comunicación de las neuronas con las células post sinápticas.
Otras mutaciones que causan defectos en el citoesqueleto están relacionadas también con la ELA, confirmando éstas como una de las causas de la enfermedad. Además, alteraciones en dos rutas metabólicas también pueden ser causas de esta patología.
Este gen, ya había sido descrito como causante de otras dos enfermedades, paraplejia espástica hereditaria (HSP) y enfermedad de Charcot-Marie tipo 2 (CMT2). Sin embargo, y a diferencia con la ELA, las mutaciones se localizan en el extremo N-terminal (base de la proteína KIF5A), mientras que en la ELA, se encuentra en el C-terminal (extremo de unión a moléculas transportadoras de la proteína).
Por tanto, el siguiente objetivo es estudiar por qué una mutación en un extremo causa ELA y en el otro extremo causan HSP para así, desarrollar fármacos contra estas enfermedades.
Noticia
https://revistageneticamedica.com/2018/04/10/citoesqueleto-y-ela/
Discovery of new ALS gene points to common role of cytoskeleton in disease. https://www.umassmed.edu/news/news-archives/2018/03/discovery-of-new-als-gene-points-to-common-role-of-cytoskeleton-in-disease/
Referencia
Nicolas A, et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron. 2018 Mar 21;97(6):1268-1283.e6. doi: 10.1016/j.neuron.2018.02.027.
By Javier Gómez (DGMol)
Retractado el artículo que afirmaba que CRISPR introduce cientos de mutaciones no deseadas en el genoma
El pasado junio investigadores de la Universidad de Columbia publicaron un artículo en Nature Methods, en el cual afirmaban que el sistema de edición genética CRISPR puede causar cientos de mutaciones no deseadas en paralelo a la introducción del cambio deseado.
Esta publicación fue criticada por la comunidad científica, se intentaron buscar las causas de estas mutaciones, ajenas al sistema CRISPR. Por otro lado, este articulo generó un gran impacto mediático, afectando a empresas biotecnológicas relacionadas con el uso de CRISPR
Nature Methods finalmente ha retractado el trabajo llevado a cabo por Schaefer, debido a que los datos proporcionados eran insuficientes para afirmar la existencia de mutaciones no deseadas asociadas a las técnicas de CRISPR, ya que es necesario más trabajos para afirmar que estas mutaciones ocurren in vivo.
La revista señala que el principal error de este estudio es que los investigadores no tuvieron en cuenta el fondo genético de los ratones utilizados. Debido a esto, no se puede afirmar que las variaciones genéticas encontradas son provocadas por CRISPR o forma parte de la composición genómica inicial. Además, la revista señala el reducido número de animales utilizados como otro error
Estudios llevados a cabo para analizar el genoma completo de embriones humanos modificados por CRISPR no han observado mutaciones colaterales a esta técnica. Por otro lado, la publicación en un nuevo trabajo depositado en bioRxiv concluye que CRISPR permite editar el genoma de forma precisa, sin provocar mutaciones colaterales. Existen multitud de trabajos que apoyan el desarrollo de CRISPR como herramienta terapéutica para corregir defectos genéticos. A pesar de esto, para usar estas técnicas es necesario realizar estudios que confirmen su seguridad y precisión en humanos, ya que utilizar CRISPR in vivo pone mucho en juego y los pacientes que usen esta técnica merecen una respuesta con rigor.
Noticia
https://revistageneticamedica.com/2018/04/05/retractado-crispr-mutaciones/
Referencias
CRISPR off-targets: a reassessment. Nature Meth. 2018. Doi: http://dx.doi.org/10.1038/nmeth.4664
Schaefer KA, et al. Corrigendum and follow-up: Whole genome sequencing of multiple CRISPR-edited mouse lines suggests no excess mutations. bioRxiv. 2018. Doi: https://doi.org/10.1101/154450
By Fco Javier Muñoz (DGMol)
Una mutación poco común en el gen EPO produce eritrocitosis hereditaria
Investigadores de la Universidad de Basilea acaban de identificar una mutación poco común en el gen EPO que lleva al desarrollo de eritrocitosis hereditaria, condición caracterizada por el aumento de eritrocitos o glóbulos rojos en la sangre.
 En un reciente estudio, publicado en el New England Journal of Medicine, los investigadores describen cómo el análisis del genoma de diversos miembros de una familia con eritrocitosis hereditaria autosómica dominante permitió relacionar una mutación en el gen EPO con la enfermedad.
La eritrocitosis hereditaria primaria con niveles bajos de eritropoyetina en sangre se produce debido a mutaciones en el receptor de la eritropoyetina (EPOR), en SH2B3 o JAK2, mientras que en los casos que cursan con niveles altos de eritropoyetina se producen principalmente debido a mutaciones en genes relacionados con la percepción del oxígeno.
Con el fin de identificar al gen responsable, el siguiente paso de los investigadores fue llevar a cabo un análisis de ligamiento que permitió acotar la región del genoma que cosegregaba con la enfermedad.
La mutación identificada por los investigadores es una deleción de un nucleótido que lleva a un cambio de pauta de lectura en el ARN mensajero de EPO. Este cambio rompe el péptido señal de la eritropoyetina y lleva a una proteína incompleta que no funciona correctamente. Así, lo esperado ante una mutación que lleva a falta de proteína funcional sería encontrar que los portadores de la mutación no tenían eritropoyetina funcional en sangre. in embargo, el equipo observó que la mayoría de los afectados de la familia tenían niveles aumentados de eritropoyetina en sangre.
Intrigados, los investigadores crearon una línea celular en la que introdujeron la mutación identificada mediante CRISPR y analizaron el ARN mensajero derivado de EPO.
Descubrieron que la mutación que trunca el transcrito principal del gen EPO e impide la producción de proteína a partir de éste, también altera un transcrito no codificante y lo convierte en uno que produce eritropoyetina en exceso y lleva al desarrollo de la enfermedad.
En un reciente estudio, publicado en el New England Journal of Medicine, los investigadores describen cómo el análisis del genoma de diversos miembros de una familia con eritrocitosis hereditaria autosómica dominante permitió relacionar una mutación en el gen EPO con la enfermedad.
La eritrocitosis hereditaria primaria con niveles bajos de eritropoyetina en sangre se produce debido a mutaciones en el receptor de la eritropoyetina (EPOR), en SH2B3 o JAK2, mientras que en los casos que cursan con niveles altos de eritropoyetina se producen principalmente debido a mutaciones en genes relacionados con la percepción del oxígeno.
Con el fin de identificar al gen responsable, el siguiente paso de los investigadores fue llevar a cabo un análisis de ligamiento que permitió acotar la región del genoma que cosegregaba con la enfermedad.
La mutación identificada por los investigadores es una deleción de un nucleótido que lleva a un cambio de pauta de lectura en el ARN mensajero de EPO. Este cambio rompe el péptido señal de la eritropoyetina y lleva a una proteína incompleta que no funciona correctamente. Así, lo esperado ante una mutación que lleva a falta de proteína funcional sería encontrar que los portadores de la mutación no tenían eritropoyetina funcional en sangre. in embargo, el equipo observó que la mayoría de los afectados de la familia tenían niveles aumentados de eritropoyetina en sangre.
Intrigados, los investigadores crearon una línea celular en la que introdujeron la mutación identificada mediante CRISPR y analizaron el ARN mensajero derivado de EPO.
Descubrieron que la mutación que trunca el transcrito principal del gen EPO e impide la producción de proteína a partir de éste, también altera un transcrito no codificante y lo convierte en uno que produce eritropoyetina en exceso y lleva al desarrollo de la enfermedad.
Noticia
https://revistageneticamedica.com/2018/03/27/eritrocitosis-hereditaria/
Inherited mutation leads to overproduction of EPO. https://www.unibas.ch/en/News-Events/News/Uni-Research/Inherited-mutation-leads-to-overproduction-of-EPO.html
Referencia
Zmajkovic J, et al. A Gain-of-Function Mutation in EPO in Familial Erythrocytosis. N Engl J Med. 2018 Mar 8;378(10):924-930. doi: http://dx.doi.org/10.1056/NEJMoa1709064
By Alicia Lavado (GMed)
CRISPR para identificar nuevas dianas terapéuticas para la esclerosis lateral amiotrófica (ELA)
Investigadores de la Universidad de Stanford han identificado nuevas dianas para el tratamiento de la esclerosis lateral amiotrófica y la demencia frontolateral mediante un sistema de rastreo molecular basado en la tecnología CRISPR de edición del genoma.
 La expansión de un fragmento del gen C9ORF72 es la causa genética más común de esclerosis lateral amiotrófica (ELA) y demencia frontolateral (DFL), enfermedades neurodegenerativas que si bien difieren en su manifestación principal, comparten características que han llevado a plantear que sean consideradas como parte de un mismo espectro clínicopatológico.
La mutación en C9ORF72 lleva a la producción de proteínas anómalas que pueden formar agregados tóxicos para las células nerviosas, e inducir la muerte neuronal y posterior aparición de los diversos síntomas neurológicos.
El objetivo de los investigadores del trabajo era identificar qué genes actúan como moduladores de la toxicidad de C9ORF72. Para ello llevaron a cabo un rastreo en el que inactivaron uno por uno los diferentes genes del genoma mediante el sistema de edición genómica CRISPR y observaron el efecto de esta inactivación en la supervivencia de las células.
Si la inactivación de un gen lleva a que aumente la supervivencia de las células, esto significa que el gen contribuye a proteger las células de la toxicidad causada por las proteínas anómalas. Este tipo de genes pueden ser utilizados en el futuro como dianas terapéuticas en el desarrollo de fármacos para tratar la ELA o la DFL. Si por el contrario la inactivación lleva a aumentar la mortalidad
celular esto significa que el gen contribuye a empeorar la situación fisiológica celular ocasionada por los agregados de proteínas. En ambos casos, los investigadores obtienen información sobre sobre los mecanismos moleculares que intervienen en la degeneración que ocurre en estas enfermedades.
El rastreo permitió identificar 200 genes que al ser inactivados contribuyen a proteger a las células del efecto tóxico de los agregados de proteínas tóxicas o hacen las células más sensibles a la toxicidad. Con el fin de reducir el número a los candidatos más probables, los investigadores validaron los resultados en cultivos primarios de neuronas de ratón. Algunos de los genes detectados están relacionados con la maquinaria molecular responsable del transporte de sustancias entre el núcleo y el citoplasma de la célula, lo que respalda los resultados de estudios anteriores. Además, los investigadores identificaron otros genes que sugieren que el retículo endoplásmico es un elemento central en la patología provocada por C9ORF72.
Dentro de los genes identificados, los investigadores prestaron especial atención al gen Tmx2. Este gen codifica para una proteína que forma parte del retículo endoplásmico e interviene en la respuesta a estímulos de estrés que pueden causar la muerte celular. “Estamos en las fases
tempranas pero creo que descubrir exactamente lo que Tmx2 hace en la célula es un buen punto en el que empezar, que nos daría pistas sobre qué funciones están alteradas cuando las formas tóxicas inducen la muerte celular y podría indicarnos qué rutas deberíamos investigar,” manifiesta Nicholas Kramer, uno de los investigadores del trabajo.
Los resultados del trabajo apuntan a posibles dianas terapéuticas para la ELA y la DFL. Sin embargo conviene tener en cuenta que la toxicidad mediada por la mutación en C9ORF72 no se limita a la producción de agregados tóxicos de proteínas, sino que también puede ser producida por las expansiones en el ARN o la pérdida de función del gen C9ORF72. Los investigadores señalan que estudios futuros deberán estimar en qué medida contribuyen cada uno de estos factores al desarrollo de la patología para poder considerar a los supresores de las proteínas tóxicas como dianas terapéuticas.
Aunque en este caso el sistema CRISPR se ha utilizado únicamente para inactivar uno por uno los genes del genoma, la tecnología puede modificarse para modular la expresión génica y llevar otro tipo de rastreos. Además, los investigadores evalúan en la actualidad si el mismo sistema podría ser utilizado para estudiar no sólo la esclerosis lateral amiotrófica o la demencia frontolateral, sino también otras enfermedades neurodegenerativas causadas por proteínas que inducen toxicidad.
La expansión de un fragmento del gen C9ORF72 es la causa genética más común de esclerosis lateral amiotrófica (ELA) y demencia frontolateral (DFL), enfermedades neurodegenerativas que si bien difieren en su manifestación principal, comparten características que han llevado a plantear que sean consideradas como parte de un mismo espectro clínicopatológico.
La mutación en C9ORF72 lleva a la producción de proteínas anómalas que pueden formar agregados tóxicos para las células nerviosas, e inducir la muerte neuronal y posterior aparición de los diversos síntomas neurológicos.
El objetivo de los investigadores del trabajo era identificar qué genes actúan como moduladores de la toxicidad de C9ORF72. Para ello llevaron a cabo un rastreo en el que inactivaron uno por uno los diferentes genes del genoma mediante el sistema de edición genómica CRISPR y observaron el efecto de esta inactivación en la supervivencia de las células.
Si la inactivación de un gen lleva a que aumente la supervivencia de las células, esto significa que el gen contribuye a proteger las células de la toxicidad causada por las proteínas anómalas. Este tipo de genes pueden ser utilizados en el futuro como dianas terapéuticas en el desarrollo de fármacos para tratar la ELA o la DFL. Si por el contrario la inactivación lleva a aumentar la mortalidad
celular esto significa que el gen contribuye a empeorar la situación fisiológica celular ocasionada por los agregados de proteínas. En ambos casos, los investigadores obtienen información sobre sobre los mecanismos moleculares que intervienen en la degeneración que ocurre en estas enfermedades.
El rastreo permitió identificar 200 genes que al ser inactivados contribuyen a proteger a las células del efecto tóxico de los agregados de proteínas tóxicas o hacen las células más sensibles a la toxicidad. Con el fin de reducir el número a los candidatos más probables, los investigadores validaron los resultados en cultivos primarios de neuronas de ratón. Algunos de los genes detectados están relacionados con la maquinaria molecular responsable del transporte de sustancias entre el núcleo y el citoplasma de la célula, lo que respalda los resultados de estudios anteriores. Además, los investigadores identificaron otros genes que sugieren que el retículo endoplásmico es un elemento central en la patología provocada por C9ORF72.
Dentro de los genes identificados, los investigadores prestaron especial atención al gen Tmx2. Este gen codifica para una proteína que forma parte del retículo endoplásmico e interviene en la respuesta a estímulos de estrés que pueden causar la muerte celular. “Estamos en las fases
tempranas pero creo que descubrir exactamente lo que Tmx2 hace en la célula es un buen punto en el que empezar, que nos daría pistas sobre qué funciones están alteradas cuando las formas tóxicas inducen la muerte celular y podría indicarnos qué rutas deberíamos investigar,” manifiesta Nicholas Kramer, uno de los investigadores del trabajo.
Los resultados del trabajo apuntan a posibles dianas terapéuticas para la ELA y la DFL. Sin embargo conviene tener en cuenta que la toxicidad mediada por la mutación en C9ORF72 no se limita a la producción de agregados tóxicos de proteínas, sino que también puede ser producida por las expansiones en el ARN o la pérdida de función del gen C9ORF72. Los investigadores señalan que estudios futuros deberán estimar en qué medida contribuyen cada uno de estos factores al desarrollo de la patología para poder considerar a los supresores de las proteínas tóxicas como dianas terapéuticas.
Aunque en este caso el sistema CRISPR se ha utilizado únicamente para inactivar uno por uno los genes del genoma, la tecnología puede modificarse para modular la expresión génica y llevar otro tipo de rastreos. Además, los investigadores evalúan en la actualidad si el mismo sistema podría ser utilizado para estudiar no sólo la esclerosis lateral amiotrófica o la demencia frontolateral, sino también otras enfermedades neurodegenerativas causadas por proteínas que inducen toxicidad.
Noticia
Potential drug targets for ALS revealed in study using CRISPR. http://med.stanford.edu/news/all-news/2018/03/potential-drug-targets-for-als-revealed-in-study-using-crispr.html
Referencia
Kramer NJ, et al. CRISPR-Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nat Genet. 2018 Mar 5. doi: http://dx.doi.org/10.1038/s41588-018-0070-7.
By Luis Vera (GMed)